I use c++ to do most of my analysis. Sometimes, I might want to do some small calculations on the results from c++, and it can be executed faster with a script. I've used bash script to get certain information in c++ output files. While bash script might be handy to use, it can only do integer arithmetic.
I have two columns of data, in which I want to manipulate the values in the second column. After realizing that bash script wouldn't do floating-point calculation for me, perl became my next option.
To be honest, I've never used it, never seen one single perl script before this evening. All the credits of my Perl script go to the forums on the web, and people who contributed to those threads.
What I wanted to do is simple, but I found it is really hard to get a direct solution by googling around. I use pieces of instructions and examples from more than 10 webpages. My hope is that by putting together what I learnt tonight, newbies to Perl would understand how to:
- read in data from a file
- and store elements in arrays
- do simple calculations
- output result
Maybe the reason that there's no direct solution is that there's a much easier way out there than using Perl. But anyway, here's the script:
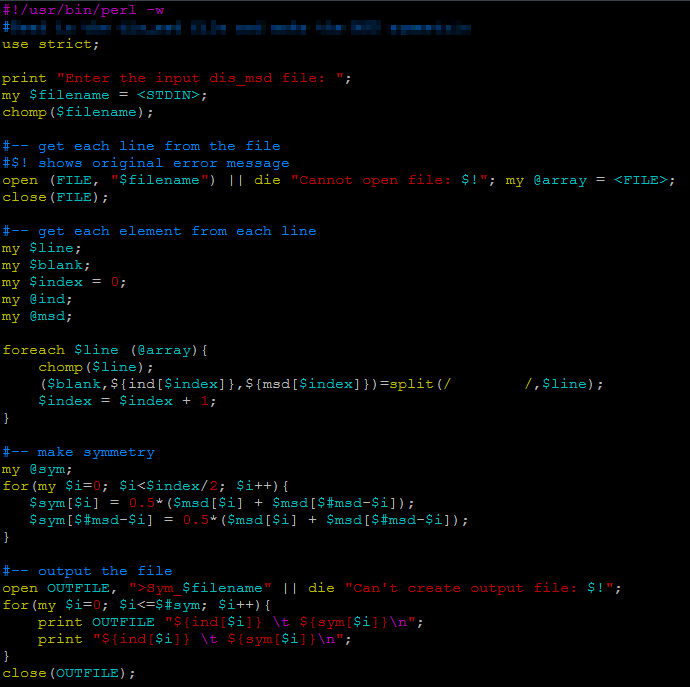
Perl is similar to c++, one big difference is that for every variable, a "$" sign need to come before it.
First ask user to input the file that will be read in:
| print "Enter the input file: "; |
| my $filename = <STDIN>; |
my is used to declare a variable locally. With the use of "use strict", if we use a variable without defining it, Perl will show an error message.
<STDIN> is what being typed by the user at the command prompt. It stands for standard input, and can be abbreviated by using simple <>. One thing you might already noticed is that unlike bash script, Perl requires a ";" at the end of a command, which is the same as c++.
After getting the input file name, we first read in each line of the file:
| open (FILE, "$filename") || die "Cannot open file: $!"; |
| my @array = <FILE>; |
| close(FILE) |
Each line in the file will be stored in the
array. In Perl, an array is defined with a
@ sign, and a scalar is defined with a
$ in front of the variable name:
line will be used to store one value.
ind will be used as an array.
Now that we have each line stored in
array, we want to extract the elements and assign them to array
@ind and
@msd:
| foreach $line (@array){ |
| chomp($line); |
| ($blank,${ind[$index]},${msd[$index]})=split(/ /,$line); |
| $index = $index + 1; |
| } |
In the
foreach loop, we first use
chomp function to remove the newline character in the string. When reading data from user or from a file, usually there's a newline character at the end of the string. My input file looks like this:
1 3.5555
2 4.0233
3 3.5099
.. .........
.. ..........
There's a huge blank space before the first column (it has to do with the way my c++ code output the results). The
split function allows you to define what the deliminator is in each line of your
array. I was able to store each column in respective array by storing part of the blank space in front in another variable
$blank, which I had no intention to use afterwards.
After storing the data into those arrays, I can do calculations with those numbers. If I would have a c++ code to do the same thing, it'd take me just a couple of minutes. But I like the convenience that scripting language could give me: execute the program without compiling it. Compiling a code meaning to add an addition executable file to the folder. If you have more than one executable, you need to name them in a reasonable fashion so that you know which one does which function. Usually, in this case I just recompile the code so that I'm certain the executable I'm using is the right one. Using scripting program not only reduces the number of files in the folder, but also eliminates the hassle of compiling codes.
Maybe the
awk function could do this a lot easier than what I'm doing here. I'll try to expand my horizon to this field .... sometime when I finish my ChemE degree at Penn State.






