Although we could always write our own scripts to process LAMMPS output files, LAMMPS has this awesome package,
Pizza.py , which already contains many powerful tools to allow quick observations on the simulation results.
At the beginning of a simulation, you might want to check how energies and temperature drift, get averages over blocks of trajectory, and visualize the fluctuation of the simulation box. All these information can be retrieved in the log file, and all we need is the Pizza.py toolkit. Since the whole package is a combination of Python scripts, we also need to have Python installed.
Download the Pizza.py toolkit from
this page , and put it in your directory. After unzipping it, you could find the Python script pizza.py in the
src folder. The Pizza.py toolkit can be used directly or interactively.
First set up an alias pointing to where you put the pizza.py file. For example:
| $
alias pizza='python -i /home/pizza/src/pizza.py
|
Then run pizza.py by typing in
This will bring you into the pizza.py interactive interface:
|
Pizza.py (9 Feb 2012), a toolkit written in Python
type ? for help, CTRL-D to quit
Loading tools ...
>
|
There might be some error messages after
Loading tools if you don't have some of the modules required. Don't worry if you are missing some modules. Not every function uses all the modules.
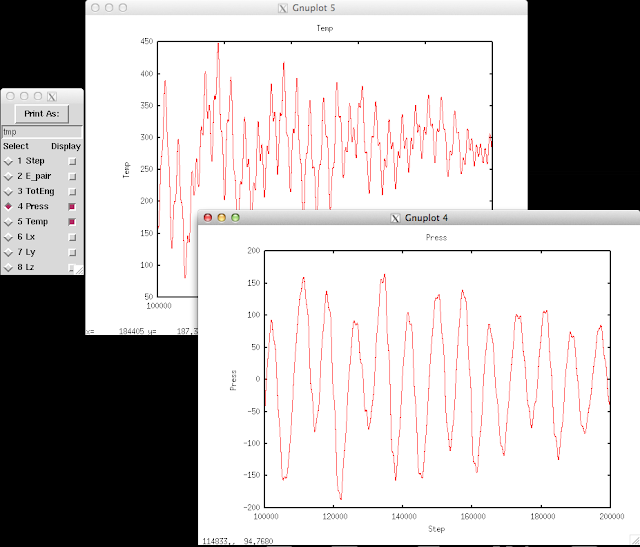
Here I want to demonstrate how to use the interactive interface to observe the thermo output in LAMMPS log file.
|
> l=log("*log")
180000000 182500000 185000000 187500000 190000000 192500000
read 6001 log entries
|
This function will read in the files that have names end with
log. This command calls the
log.py script in the
src folder. It can take multiple files, single files, even incomplete files. Check the comments in
log.py to get more information.
After reading in the thermo info stored in *log, we can get individual items. For example, to get timestep, pressure, temperature, energy:
|
s,t,p,E=l.get("Step","Temp","Press","TotEng")
|
Variables on the left hand side can just be any variables. The items inside the right hand parenthesis have to match with their names in the *log files.
The thermo information fluctuates a lot. A better way to look at them is to plot against time. In the
Pizza.py toolkit, they incorporate
Gnuplot (in
gnu.py) so that we can directly use it in the interactive interface:
The first command calls Gnuplot and the second command will plot timesteps vs. temperature.
If you are like me, impatient of typing in those commands repetitively, don't worry. Pizza.py toolkit can also take scripts. There is
a collection of scripts written by Pizza.py users. They come within the Pizza.py package. These scripts are handy to use and are very powerful. It allows me to do block averages in just one line.
Read more:
Block average
Visualizing log files
 (About how to use Pizza.py, please refers to previous post of How to use Pizza.py for LAMMPS or the official Pizza.py Toolkit page.
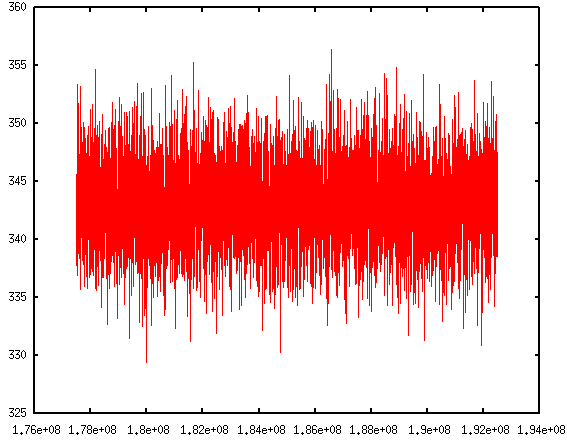
Instead of asking pizza.py to get all the "Temp", "TotEng" .. by typing in each variable, and then do g.plot (a lot of repetitive keyboard work!), there's a much faster way to visualize the thermo-type data. It allows us to quickly view the trend of properties, and get an idea of the equilibrium status of the simulation.
This can be done directly on the shell or through pizza.py. Below is to do it in pizza.py:
(About how to use Pizza.py, please refers to previous post of How to use Pizza.py for LAMMPS or the official Pizza.py Toolkit page.
Instead of asking pizza.py to get all the "Temp", "TotEng" .. by typing in each variable, and then do g.plot (a lot of repetitive keyboard work!), there's a much faster way to visualize the thermo-type data. It allows us to quickly view the trend of properties, and get an idea of the equilibrium status of the simulation.
This can be done directly on the shell or through pizza.py. Below is to do it in pizza.py: